
Il cancro batterico dell'actinidia causato da Pseudomonas syringae pv. actinidiae (Psa) è un esempio emblematico di una patologia particolarmente dannosa che sta devastando la filiera di una frutticola molto importante come il kiwi sia a livello nazionale, sia mondiale.
Dal 2008, Psa si è manifestato e diffuso nella sua forma più virulenta devastando migliaia di ettari di Actinidia spp. in tutto il mondo. Per comprendere e distinguere le differenze tra le popolazioni di Psa all'interno di questa patovar e chiarire la loro diffusione su scala mondiale, come poterle identificare e circoscriverle tempestivamente, è necessario essere in grado di confrontare in maniera approfondita e rapida le precedenti popolazioni di Psa rispetto ai casi recenti.
Il gruppo di fitobatteriolgia del Dafne, dell'Università della Tuscia negli ultimi anni ha sviluppato in questo senso un'ampia collaborazione tra Italia (Università dalla Tuscia, Dafne), Francia (Institute for integrative biology of the cell, Cnrs, Université Paris-Sud, Université Paris-Saclay, Orsay e Ensta ParisTech, Université Paris-Saclay, Palaiseau) e Nuova Zelanda (Department of biochemistry, University of Otago, Dunedin, New Zealand,), rendendo pubblici nelle scorse settimane i risultati mediante la rivista scientifica internazionale PlosOne (Development of a multiple loci variable number of tandem repeats analysis (Mlva) to Unravel the intra-pathovar structure of Pseudomonas syringae pv. actinidiae Populations Worldwide, Autori: Serena Ciarroni, Lorenzo Gallipoli, Maria C. Taratufolo, Margi I. Butler, Russell T.M., Poulter, Christine Pourcel, Gilles Vergnaud, Giorgio M. Balestra, Angelo Mazzaglia, http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0135310) e, l'intera ricerca, è stata possibile in virtù di una fattiva collaborazione con il Servizio fitosanitario centrale (Sfc) del Mipaaf e del Servizio fitosanitario regionale (Sfr) del Lazio.
Lo studio si è sviluppato applicando su una collezione di livello mondiale di 142 ceppi di Psa, la tecnica denominata Mlva (Multiple loci variable number of tandem repeats analysis). Questa tecnica, rispetto a quanto applicato fino ad oggi, è risultata maggiormente efficace, riproducibile, semplice e a basso costo. Un gruppo specifico di 13 loci Vntr è stato identificato nel genoma di Psa e quindi usato per riconoscere le differenti popolazioni del patogeno fornendo risultati particolarmente attendibili ed arrivando a distinguere ben 58 aplotipi diversi. La recente popolazione ipervirulenta di Psa mostra una diversità limitata e comprende, oltre ai ceppi provenienti da Europa, Nuova Zelanda e Cile, alcuni ceppi della regione dello Shaanxi, in Cina. Un'ampia variabilità genetica è stata osservata per gli isolati di Psa ottenuti in Cina, ma differenti subpopolazioni sono state distinte e caratterizzate anche per quanto riguarda le popolazioni di Psa ad oggi originarie del Giappone e della Corea.
I ceppi di Psa a ridotta virulenza sono risultati distinti e raggruppati tra di loro e sono risultati molto differenti dagli altri genotipi evidenziati mediante l'analisi Mlva.
Questi dati hanno permesso di generare una banca dati pubblica che oggi permette di effettuare analisi di routine dei loci Vntr presenti nel genoma di Psa ed ottenere precise indicazioni sulla appartenenza a specifiche popolazioni del patogeno riducendo significativamente i tempi, i costi e fornendo risposte di assoluta attendibilità.
I risultati dello studio evidenziano che, rispetto alle 4 popolazioni (biovar) di Psa note, oggi in realtà siamo in presenza di almeno 10 popolazioni di Psa su scala mondiale.
Poter identificare con tempestività e precisione le differenti popolazioni di Psa risulta di fondamentale importanza sia per interrompere/limitare la diffusione del batterio su scala mondiale, come per circoscrivere eventuali focolai e, mediante questi risultati, la ricerca italiana fornisce un'ulteriore contributo a sostegno di un prodotto agroalimentare made in Italy di eccellenza.
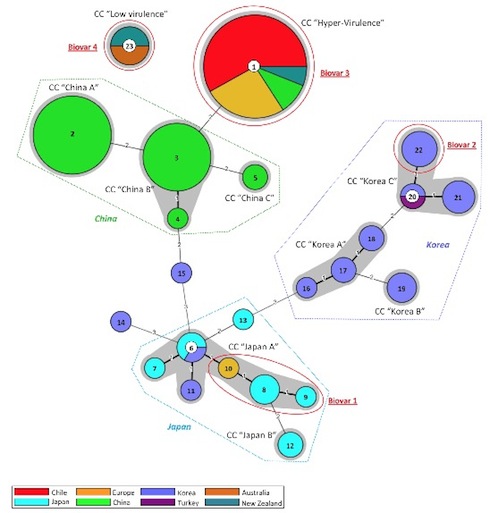
Le popolazioni di Psa a livello mondiale: risultati ottenuti mediante analisi Mlva
Minimum spanning tree (Mst) che riproduce graficamente le relazioni tra le diverse popolazioni di Psa. Ogni cerchio rappresenta un gruppo di ceppi identici tra di loro (aplotipo) e le dimensioni di ogni cerchio sono proporzionali al numero di isolati in esso contenuti. Ciascuna area colorata di grigio rappresenta un complesso clonale (CC) ovvero gruppi di isolati di Psa che differiscono tra loro al massimo in 1 locus su 13. I cerchi rossi racchiudono i ceppi di Psa appartenenti alle 4 biovars riconosciute fino ad oggi.
Dal 2008, Psa si è manifestato e diffuso nella sua forma più virulenta devastando migliaia di ettari di Actinidia spp. in tutto il mondo. Per comprendere e distinguere le differenze tra le popolazioni di Psa all'interno di questa patovar e chiarire la loro diffusione su scala mondiale, come poterle identificare e circoscriverle tempestivamente, è necessario essere in grado di confrontare in maniera approfondita e rapida le precedenti popolazioni di Psa rispetto ai casi recenti.
Il gruppo di fitobatteriolgia del Dafne, dell'Università della Tuscia negli ultimi anni ha sviluppato in questo senso un'ampia collaborazione tra Italia (Università dalla Tuscia, Dafne), Francia (Institute for integrative biology of the cell, Cnrs, Université Paris-Sud, Université Paris-Saclay, Orsay e Ensta ParisTech, Université Paris-Saclay, Palaiseau) e Nuova Zelanda (Department of biochemistry, University of Otago, Dunedin, New Zealand,), rendendo pubblici nelle scorse settimane i risultati mediante la rivista scientifica internazionale PlosOne (Development of a multiple loci variable number of tandem repeats analysis (Mlva) to Unravel the intra-pathovar structure of Pseudomonas syringae pv. actinidiae Populations Worldwide, Autori: Serena Ciarroni, Lorenzo Gallipoli, Maria C. Taratufolo, Margi I. Butler, Russell T.M., Poulter, Christine Pourcel, Gilles Vergnaud, Giorgio M. Balestra, Angelo Mazzaglia, http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0135310) e, l'intera ricerca, è stata possibile in virtù di una fattiva collaborazione con il Servizio fitosanitario centrale (Sfc) del Mipaaf e del Servizio fitosanitario regionale (Sfr) del Lazio.
Lo studio si è sviluppato applicando su una collezione di livello mondiale di 142 ceppi di Psa, la tecnica denominata Mlva (Multiple loci variable number of tandem repeats analysis). Questa tecnica, rispetto a quanto applicato fino ad oggi, è risultata maggiormente efficace, riproducibile, semplice e a basso costo. Un gruppo specifico di 13 loci Vntr è stato identificato nel genoma di Psa e quindi usato per riconoscere le differenti popolazioni del patogeno fornendo risultati particolarmente attendibili ed arrivando a distinguere ben 58 aplotipi diversi. La recente popolazione ipervirulenta di Psa mostra una diversità limitata e comprende, oltre ai ceppi provenienti da Europa, Nuova Zelanda e Cile, alcuni ceppi della regione dello Shaanxi, in Cina. Un'ampia variabilità genetica è stata osservata per gli isolati di Psa ottenuti in Cina, ma differenti subpopolazioni sono state distinte e caratterizzate anche per quanto riguarda le popolazioni di Psa ad oggi originarie del Giappone e della Corea.
I ceppi di Psa a ridotta virulenza sono risultati distinti e raggruppati tra di loro e sono risultati molto differenti dagli altri genotipi evidenziati mediante l'analisi Mlva.
Questi dati hanno permesso di generare una banca dati pubblica che oggi permette di effettuare analisi di routine dei loci Vntr presenti nel genoma di Psa ed ottenere precise indicazioni sulla appartenenza a specifiche popolazioni del patogeno riducendo significativamente i tempi, i costi e fornendo risposte di assoluta attendibilità.
I risultati dello studio evidenziano che, rispetto alle 4 popolazioni (biovar) di Psa note, oggi in realtà siamo in presenza di almeno 10 popolazioni di Psa su scala mondiale.
Poter identificare con tempestività e precisione le differenti popolazioni di Psa risulta di fondamentale importanza sia per interrompere/limitare la diffusione del batterio su scala mondiale, come per circoscrivere eventuali focolai e, mediante questi risultati, la ricerca italiana fornisce un'ulteriore contributo a sostegno di un prodotto agroalimentare made in Italy di eccellenza.
Le popolazioni di Psa a livello mondiale: risultati ottenuti mediante analisi Mlva
Minimum spanning tree (Mst) che riproduce graficamente le relazioni tra le diverse popolazioni di Psa. Ogni cerchio rappresenta un gruppo di ceppi identici tra di loro (aplotipo) e le dimensioni di ogni cerchio sono proporzionali al numero di isolati in esso contenuti. Ciascuna area colorata di grigio rappresenta un complesso clonale (CC) ovvero gruppi di isolati di Psa che differiscono tra loro al massimo in 1 locus su 13. I cerchi rossi racchiudono i ceppi di Psa appartenenti alle 4 biovars riconosciute fino ad oggi.
© AgroNotizie - riproduzione riservata
Fonte: Università della Tuscia




















